Zunächst zu der als Late-breaking Abstract von Philip J. Mease, Seattle (USA), und seinen internationalen Kollegen vorgestellten randomisierten, doppelblinden, placebokontrollierten Phase-II-Studie zur Effektivität und Sicherheit des humanisierten monoklonalen IgG1-Antikörpers und selektiven IL-23p19-Inhibitors Risankizumab. Hierin wurden 185 Patienten mit aktiver PsA (im Mittel 51 Jahre, 43 % Frauen, 49 % mit Psoriasis ≥3% KOF, 30 bzw. 65 % mit Daktylitis/Enthesitis) im Verhältnis 2:2:2:1:2 auf Risankizumab 150 mg in Woche 0, 4, 8, 12 und 16 (Arm 1), 150 mg in Woche 0, 4 und 16 (Arm 2), 150 mg in Woche 0 und 12 (Arm 3), 75 mg als Einzeldosis in Woche 0 (Arm 4) oder Placebo (Arm 5) randomisiert. Vorab war eine Stratifizierung nach früherem Anti-TNF- (24,3 %) und begleitendem Methotrexat (MTX)-Gebrauch (57,3 %) erfolgt. Primärer Wirksamkeits-Endpunkt war das ACR20-Ansprechen in Woche 16.
Anti-IL-23: Phase-II-Daten zu Risankizumab und Guselkumab
In Woche 16 zeigte sich über alle Risankizumab-Arme hinweg ein signifikant besseres ACR20-Ansprechen (57,1-65,0 %) gegenüber Placebo (37,5 %). Meist nur numerisch besser war das ACR50-Ansprechen (23,8-38,5 %), vielfach signifikant überlegen waren das ACR70-Ansprechen (7,1-25,6 %), das Erreichen einer minimalen Krankheitsaktivität (MDA) (28,6-35,0 %), der ΔDAS28-CRP (max. -1,9) und die Schmerzreduktion (max. -24,3). Erwartungsgemäß war das PASI75/90/100-Ansprechen (66,7-75,0 %/52,2-66,7 %/33,3-55,6 %) in Woche 16 unter Risankizumab fast durchweg signifikant höher im Vergleich zu Placebo. Nur numerische Vorteile waren aber in puncto HAQ-DI, Enthesitis und Daktylitis zu verzeichnen. Therapie-assoziierte unerwünschte Ereignisse (UE) traten in allen Armen vergleichbar selten auf (am häufigsten Infektionen). In dieser Phase-II-Studie verbesserte Risankizumab vorrangig die Haut- aber durchaus auch klinisch relevant die Gelenksymptomatik bei guter Verträglichkeit. Jedoch wurde aufgrund der geringen Gruppengrößen vielfach keine statistische Signifikanz erreicht, auch ergibt sich (noch) kein klares Bild bezüglich der besten Dosis und Applikationsfrequenz. (1)
Etwas weiter scheint mit Guselkumab der erste in einer randomisierten, doppelblinden, placebokontrollierten Phase-IIa-Studie bei PsA getestete reine IL-23-Hemmer zu sein, dies jedoch in einem klarer umrissenen Kollektiv von trotz einer Standardtherapie mit DMARDs und z. T. auch TNFα-Inhibitoren aktiven PsA-Patienten (SJC/TJC ≥3, CRP ≥3 mg/l), die von vornherein eine Psoriasis (≥3 % KOF) aufweisen mussten. Zunächst waren 149 Patienten für 24 Wochen 2:1 auf Guselkumab 100 mg s.c. (Woche 0 und 4, danach alle 8 Wochen) oder Placebo randomisiert worden (bei Ineffektivität in Woche 16 „early escape“ auf Ustekinumab). In Woche 24 – alle primären und sekundären Endpunkte waren signifikant erreicht worden – wechselten dann alle übrigen Placebo-Patienten auf Verum bis Woche 44 mit einem Follow-up bis Woche 56. Über den Therapieverlauf der Patienten ohne early escape von Woche 24 bis 56 berichteten nun Atul A. Deodhar, Portland (USA), und Kollegen.
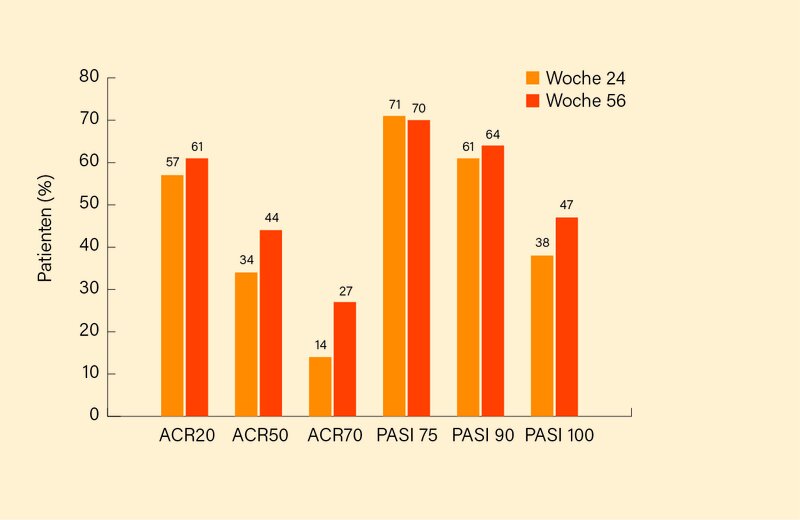
In Woche 24 wechselten 29 der ursprünglich 49 Placebo-Patienten auf Guselkumab, auf dem IL-23-Hemmer blieben 86 der zu Beginn 100 Teilnehmer. Ab Woche 24 kam es in Woche 44 und 56 zu einer deutlichen Verbesserung der ACR- und PASI-Response bei den von Placebo auf Guselkumab gewechselten Patienten: So betrug z. B. das ACR20/50/70- Ansprechen in Woche 56 81,5, 66,7 und 28,6 %, das PASI75/90/100-Ansprechen 81,5, 74,1 und 55,6 %. Im Fall der durchgehend mit Guselkumab behandelten Patienten wurden die in Woche 24 erzielten Ergebnisse aufrechterhalten oder noch verbessert auf (Woche 56) für das ACR20/50/70-Kriterium 73,5, 53,0 und 32,5 % und für das PASI75/90/100-Ansprechen 85,4, 78,0 und 57,3 % (Abb. 1). Der Anteil von Patienten mit MDA stieg von 26,7 (Woche 24) auf 34,5 % (Woche 44) an, der Anteil Patienten mit nicht völlig zurückgedrängter Enthesitis bzw. Daktylitis fiel (von Woche 24 bis 56) von 38,8 auf 29,2 % bzw. von 40,0 auf 25,5 % ab. Generell war nur eine geringe Therapiedifferenz zwischen Woche 44 (letzte Guselkumab-Applikation) und dem „off drug“-Follow-up in Woche 56 erkennbar. Die Verträglichkeit war auch über 56 Wochen gut (nur sechs schwere UE) mit keinen neuen Sicherheitssignalen.
Somit zeigte Guselkumab in diesem PsA-Kollektiv über ein Jahr hinweg eine fortgesetzte substanzielle Verbesserung von Gelenken, Haut, Enthesitis und Daktylitis sowie körperlicher Funktion und Lebensqualität – auf die weitere Evaluation in Phase-III kann man gespannt sein. (2)
Anti-IL-17A: Aktuelle Daten zu Secukinumab und Ixekizumab
Zu dem bereits zugelassenen IL-17A-Inhibitor Secukinumab wurden von Philip J. Mease, Seattle (USA), und Kollegen die Daten aus FUTURE 5, der bislang größten randomisierten, doppelblinden, placebokontrollierten Phase-III-Studie zu einem Biologikum bei PsA als Late-breaker präsentiert. Untersucht wurden darin die Effektivität einschließlich der Hemmung der röntgenologischen Progression und Sicherheit von Secukinumab 300 und 150 mg s.c. mit und – von der FDA gewünscht – ohne initiale Sättigungsdosis. Stratifiziert nach früherem Anti-TNF-Gebrauch (ca. 30 %) wurden 996 Erwachsene mit aktiver PsA im Verhältnis 2:2:2:3 auf s.c. Secukinumab 300 mg mit Sättigungsdosis (SD), 150 mg mit SD, 150 mg ohne SD oder Placebo randomisiert. Alle Gruppen erhielten Secukinumab oder Placebo zu Baseline, Woche 1, 2, 3 und 4 und danach alle 4 Wochen. In Woche 16 wechselten Placebo-Non-Responder (<20 % Verbesserung im SJC/TJC) auf Secukinumab 300 oder 150 mg, in Woche 24 dann auch die übrigen Placebo-Patienten. Der primäre Endpunkt war das ACR20-Ansprechen in Woche 16, der wichtigste sekundäre Endpunkt die röntgenologische Progression struktureller Gelenkschäden (Hände, Handgelenke, Füße) im modifizierten Total van der Heijde/Sharp-Score (mTSS) in Woche 24 (alle Analysen mit Non-Responder Imputation, NRI).
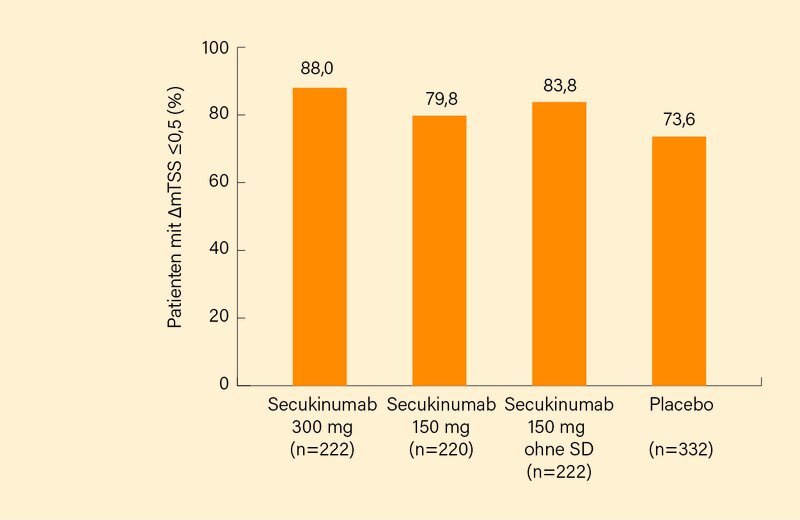
Der primäre Endpunkt des ACR20-Ansprechens in Woche 16 wurde mit allen Secukinumab-Dosierungen (ohne relevanten Unterschieden zwischen diesen) signifikant versus Placebo erreicht, gleiches galt vor allem auch für die Hemmung der Röntgenprogression im mTSS in Woche 24. Ein größerer Anteil von Patienten zeigte keine Röntgenprogression (ΔmTSS ≤0,5) unter Secukinumab gegenüber Placebo: 88 % (300 mg), 79 % (150 mg), 83 % (150 mg ohne SD) versus 73 % (Placebo) (Abb. 2). Auch alle weiteren Endpunkte wiesen signifikante Vorteile von Secukinumab versus Placebo in Woche 16 aus mit Ausnahme einer völligen Resolution von Enthesitis und Daktylitis unter Secukinumab 150 mg ohne Sättigungsdosis. Generell zeigten Patienten der 300- und 150 mg-Gruppe ein rascheres Ansprechen gegenüber jenen mit 150 mg ohne vorherige Applikation einer Sättigungsdosis.
Im Ergebnis zeigten sowohl s.c. Secukinumab 300 als auch 150 mg mit s.c.-Sättigungsdosis eine Hemmung der Röntgenprogression – zuvor war dies in FUTURE 1 streng genommen nur für die i.v.-Aufsättigung gezeigt worden – und rasche, klinisch signifikante Verbesserungen der Zeichen, Symptome und körperlichen Funktion der PsA-Patienten bei guter Verträglichkeit und ohne neue Sicherheitssignale. (3)
Unmittelbar vor der Zulassung steht nach zwei positiven Phase-III-Studien mit Ixekizumab ein zweiter IL-17A-Hemmer. Zu diesem wurden von Mark C. Genovese, Palo Alto (USA), und Kollegen die 52-Wochen-Daten der Extensionsphase der randomisierten, doppelblinden, placebokontrollierten Phase-III-Studie SPIRIT-P2 zu ursprünglich 363 PsA-Patienten mit inadäquatem Ansprechen (oder Intoleranz) auf ein oder zwei TNFα-Inhibitoren vorgestellt. In der doppelblinden Phase (Woche 0-24) hatten diese 1:1:1 s.c. 80 mg Ixekizumab alle 4 (Q4W) oder 2 Wochen (Q2W) nach einer 160 mg-Sättigungsdosis in Woche 0 oder Placebo erhalten. In der Extensionsphase (Woche 24-156) wurden 310 Patienten auf der initialen Ixekizumab-Dosis weitergeführt oder die Placebo-Patienten (Woche 16/24) re-randomisiert. In der Interimsanalyse wurden die Effektivität und Sicherheit bis Woche 52 bzw. 156 erfasst.
Den primären Endpunkt (ACR20-Ansprechen) in Woche 24 hatten signifikant mehr Patienten unter Ixekizumab Q4W (53 %) und Q2W (48 %) gegenüber Placebo (20 %) erreicht. In der Extensionsphase stieg dieses bei durchgehend damit behandelten Patienten in Woche 52 auf 68 bzw. 59 % an. Auch die ursprünglichen Placebo-Patienten zeigten einen deutlichen Benefit im ACR20 (61 bzw. 50 %) und weiteren Endpunkten. Durchweg fällt auf, dass die 80 mg Q4W-Dosierung (wie oft schon in der kontrollierten Studienphase) die etwas besseren Ergebnisse lieferte. Die von Beginn an alle 4 Wochen mit Ixekizumab 80 mg behandelten Patienten erreichten z. B. in Woche 52 ein ACR50/70-Ansprechen zu 46 bzw. 29 %, eine MDA in
38 % der Fälle, einen PASI75/90 zu 66 bzw. 55 % und 53 respektive 81 % waren frei von Enthesitis bzw. Daktylitis. Die meisten UE bis Woche 156 waren nur mild oder moderat ausgeprägt, schwere UE traten nur bei 15 Patienten auf. Alles in Allem führte Ixekizumab in diesem schwierigen Patientenkollektiv zu einer anhaltenden Verbesserung (Woche 52) der PsA-Zeichen und -Symptome bei einem zugleich über drei Jahre guten Sicherheitsprofil. (4)
TNFα-Inhibition: Golimumab i.v. sehr effektiv
Abschließend sei noch auf die von Arthur Kavanaugh, San Diego (USA), und Kollegen vorgestellte randomisierte, doppelblinde, placebokontrollierte Phase-III-Studie GO-VIBRANT zu i.v. Golimumab über 24 Wochen bei 480 Biologika-naiven Erwachsenen mit aktiver PsA verwiesen. Diese wurden 1:1 auf i.v. Golimumab 2 mg/kg in Woche 0 und 4 und danach alle 8 Wochen oder Placebo (mit Wechsel auf Golimumab in Woche 24) randomisiert. Primärer Endpunkt war das ACR20-Ansprechen in Woche 14.
Der primäre und auch alle sekundären Endpunkte wurden signifikant erfüllt. So erreichten in Woche 14 unter Golimumab gegenüber Placebo 75,1 vs. 21,8 % ein ACR20-Ansprechen – der Unterschied war bereits ab Woche 2 signifikant (45,6 vs. 7,5 %; p<0,001). Auch im ΔHAQ-DI (-0,60 vs. -0,12), ACR50 (43,6 vs. 6,3 %), ACR70 (24,5 vs. 2,1 %), PASI75 (59,2 vs. 13,6 %), MDA (27,0 vs. 4,2 %) und den Enthesitis- und Daktylitis-Scores (Δ-1,8 vs. -0,8 bzw. Δ-7,8 vs. -2,8) zeigten sich jeweils signifikante Vorteile in Woche 14 (je p<0,001). In Woche 24 erreichten überdies signifikant mehr Patienten unter Golimumab i.v. ein ACR50-Ansprechen (53,5 vs. 6,3 %) und es kam auch zu einer geringeren Progression struktureller Gelenkschäden im mTSS (Δ-0,36 vs. 1,95; je p<0,001). Das Sicherheitsprofil bis Woche 24 war vergleichbar mit jenem anderer Anti-TNF-Therapien einschließlich s.c. Golimumab, die Rate von (stets milden) Reaktionen an der Einstichstelle war gering (<2 %). Für geeignete Patienten mit aktiver PsA wäre somit die i.v.-Applikation eine mindestens ebenso effektive Alternative zur üblichen s.c.-Injektion. (5)
Quellen:
- ACR-Kongress 2017; Late-breaking Abstract 2L
- Arthritis Rheumatol 2017; 69(S10): Abstr. 2878
- ACR-Kongress 2017; Late-breaking Abstract 17L
- Arthritis Rheumatol 2017; 69(S10): Abstr. 2969
- Arthritis Rheumatol 2017; 69(S10): Abstr. 599