
: A-512 / B-418 / C-414")
Dies gelingt mit einem vielschichtigen Informationssystem, dass auf Vertragsärzte niederprasselt bis hin zum Setzen von Anreizen wie Biosimilar-Verordnungen in Verträgen zur besonderen Versorgung, der stetig zunehmenden Ausgestaltung regionaler Prüfungsvereinbarungen mit Quotenanteilen für Biosimilars und hin zur Überlegung, die Austauschpflicht für Biosimilars einzuführen.
Fraglich ist, ob in diesem Zusammenhang das Patienteninteresse gewährt bleibt und inwieweit tatsächlich ein Umstellungszwang – der zumindest so empfunden wird – gegeben ist und ob Regressgefahr droht, wenn nicht sämtliche Patienten sofort umgestellt werden. Biosimilars decken inzwischen schon über ein Drittel der Versorgung ab, die Kostenträger sparen einen dreistelligen Millionenbetrag!
EU-Zulassungen von Biosimilars
Durch die Implementierung verschiedener EU-Richtlinien (z. B. Directive 2001/83/EG) und weiterer Bestimmungen für die Zulassung von Biosimilars steht die EU hier in einer Vorreiterrolle und wird zu einem attraktiven Markt. Es gibt mehr Zulassungen als z. B. in den USA oder Japan, zumal auch die European Medicines Agency (EMA) für die Zulassung ist.
Erleichternd im Rahmen der Zulassung ist eine sog. bezugnehmende Zulassung eines bereits zugelassenen Wirkstoffs (Originator). Obwohl sich erhebliche Unterschiede zu herkömmlichen chemisch-synthetischen Arzneimitteln ergeben, da die Makromoleküle nicht nur durch ihre chemische Grundstruktur, sondern auch durch ihre räumliche Anordnung ihre biologische Aktivität entfalten, gibt es das Prinzip der Extrapolation auf die Daten des Originators, soweit in einer sensitiven Indikation ein Wirksamkeitsnachweis als Ergebnis geeigneter vorklinischer oder klinischer Versuche erbracht wurde. Die Zulassung der Biosimilars erstreckt sich somit auch auf Indikationen, die in klinischen Prüfungen des Präparates nicht überprüft wurden.
Als Folge der Extrapolation kann sich die Zulassung möglicherweise auch auf Indikationen erstrecken, für die der Originator erst später eine Zulassung erhält. Diese gelockerte Zulassungspraxis korrespondiert allerdings mit strengen Pharmakovigilanz-Auflagen. Nach Richtlinie Nummer 520/212 der EU-Kommission ist der Vertrieb von Arzneimitteln mit neuen Wirkstoffen nach der Markteinführung genauer zu beobachten. Seit dem 01.01.2014 gilt, dass die Packungen mit besonderen Kennzeichnungen zu versehen sind (sog. schwarze Triangel, also einem auf der Spitze stehendem schwarzen gleichschenkligen Dreieck). Das Symbol ist mit dem Zusatz zu versehen: „dieses Arzneimittel unterliegt einer zusätzlichen Überwachung“. Diese Pharmakovigilanz-Auflage gilt während der ersten fünf Jahre nach Marktzulassung.
Ausgehend von dieser Auflage stellt sich die Frage, ob ein Apotheker eine Substitution vornehmen darf. Die Substitutionsregelungen sind in § 129 SGB V niedergelegt und haben zur Folge, dass die Substitution maßgeblich das Marktgeschehen bei der Abgabe chemisch-synthetischer Arzneimittel steuert. Teils gibt es Rabatte von 90 % und mehr auf gelistete Herstellerabgabepreise, damit ist die aut idem-Substitution infolge der Rabattverträge aus Sicht der Krankenkassen ein effizientes Instrument zur Kostendämpfung.
Diese Regelungen sind aber nicht auf Biosimilars zu übertragen, weil es sich hierbei nicht um Biogenerika, sondern nur um ähnliche Wirkstoffe handelt, also Biosimilars.
Nach § 16 der Verfahrensordnung des Gemeinsamen Bundesausschusses (G-BA) zu Biosimilars gelten die auf biotechnologischem Weg hergestellten Präparate als derselbe Wirkstoff, soweit sie im Falle von Proteinen dieselbe Aminosäuresequenz aufweisen; dabei kann sich das Molekül in der Glykosylierung oder Tertiärstruktur unterscheiden. Unerheblich sind dabei verschiedene Salze, Ester, Ether, Isomere, Mischungen von Isomeren, Komplexe oder Derivate eines Wirkstoffes sowie nach unterschiedlichen biotechnologischen Verfahren hergestellten Produkten. Diese können nach der Grundsatzentscheidung des G-BA zu Somatropin vom März 2009 in eine einheitliche Festbetragsgruppe einfließen.
Substitution und Pharmakovigilanz
Der Substitution stehen derzeit noch Pharmakovigilanz-Auflagen entgegen, soweit gemäß § 62 Abs. 2 AMG bei der Verordnung der Name des Arzneimittels und die Nummer der Herstellungscharge genau angegeben werden sollen. Damit ist eine zweifelsfreie Produktzuordnung im Nachgang zum Auftritt von unerwünschten Arzneiwirkungen möglich, allerdings ist eine zweifelsfreie Produktzuordnung in der ärztlichen Dokumentation oft nicht gegeben.
Damit setzen sich Ärzte grundsätzlich Haftungsansprüchen aus, soweit eine mangelhafte Therapiekontrolle als Behandlungsfehler eingestuft wird.
Inzwischen ist das Gesetz für mehr Sicherheit in der Arzneimittelversorgung (GSAV) im Juli 2019 in Kraft getreten. Biosimilars sollen danach schneller in die Versorgung kommen, so dass Biosimilars, für die der G-BA eine Austauschbarkeit festgestellt hat, künftig den gleichen Austauschregelungen wie Generika unterliegen. Wenn der G-BA feststellt, dass die betreffenden Biologika austauschbar sind und der Arzt kein aut idem-Kreuz gesetzt hat, wäre die Apotheke zur Abgabe eines preisgünstigeren Biosimilars verpflichtet.
Allerdings wurde für das Inkrafttreten des aut idem-Austausches auf Apothekerebene eine Vorlaufzeit von drei Jahren festgelegt, um mehr Erfahrung im Hinblick auf die verstärkte Austauschbarkeit der Biologika zu erhalten und Verfahrensregelungen zur Beachtung der Pharmakovigilanz-Auflagen und der Dokumentation der Chargennummern zu gewährleisten. Bis zum Juli 2022 gilt somit die bisherige Rechtslage!
Der Deutsche Ärztetag hatte am 30.05.2019 eine aut idem-Substitution für Biologika ausdrücklich abgelehnt. Hingegen ergibt sich aus dem Leitfaden der Arzneimittelkommission der deutschen Ärzteschaft (AkdÄ) zum Einsatz von Biosimilars vom August 2017, dass sie sowohl bei der Erstverordnung von Biologika als auch bei der Folgeverordnung zur Fortsetzung der Therapie die jeweils wirtschaftlichere Verordnungsalternative empfiehlt.
Allerdings betont die AkdÄ die Wichtigkeit der Pharmakovigilanz und die Notwendigkeit, beobachtete Nebenwirkungen zu melden. Außerdem sei die ausführliche Patienteninformation und -beratung durch den Arzt wesentliche Voraussetzung für die Verordnung beziehungsweise den Einsatz von Biosimilars, weil andernfalls Ängste zur Schwächung der Adhärenz führen und den therapeutischen Erfolg gefährden.
Die Leitlinie „gute Substitutionspraxis“ der Deutschen Pharmazeutischen Gesellschaft hingegen lehnt bei Patienten in der Dauermedikation einen Austausch ab. Da ein Therapieerfolg bzw. ein Therapieversagen bei dieser Wirkstoffklasse nur aufwendig und in längeren Abständen überprüft werden könne, müssten sich die Betroffenen bestmöglich auf ihre Therapie verlassen können. Da ein Präparatewechsel zu einer erheblichen Verschlechterung des Gesundheitszustands führen kann, sollte im Rahmen einer Dauertherapie dringend von einem Austausch abgesehen werden.
Nach neueren Erkenntnissen des Marktforschungsinstitutes IQVIA wurde am Beispiel von Etanercept eine Erfolgsquote von ca. 80 % ermittelt, 10 % hätten zu einem Original zurückwechseln müssen, 14 % wären zu einem anderen Biologikum gewechselt (Ärztezeitung vom 25.05.2018). In rechtlicher Hinsicht bleibt der Arzt für die Therapie und die Therapiekontrolle verantwortlich.
Stellt der Arzt fest, dass trotz guter Compliance der Therapieerfolg ausbleibt oder vermeidbare Arzneimittelrisiken auftreten, sollte keine Umstellung erfolgen. Haftungsrechtlich wird eine Verteidigung mit dem Hinweis auf das Wirtschaftlichkeitsgebot nicht akzeptiert. Haftungsrechtlich kann somit eine Umstellung nur dann vorgenommen werden, wenn der Arzt sicher weiß, dass das Biosimilar in gleicher Weise wirkt. Insoweit existiert keine gesetzliche Handhabe, dem Arzt eine bestimmte Verordnungsweise (Umstellung) vorzuschreiben. Der Arzt darf die Umstellung nur vornehmen, wenn auf den ersten Blick die positiven Voraussetzungen dafür vorliegen und keine medizinischen Gründe dagegensprechen.
Quoten und Therapiefreiheit
Nach neueren Erkenntnissen beeinflusst aber auch die genetische Situation des Patienten den Erfolg einer Biologika-Therapie. So hat sich z. B. die Kombination von Methotrexat (MTX) mit dem Biologikum Adalimumab in der OPTIMA-Studie als gute Wahl in der Therapie von Patienten mit frühen Stadien der rheumatoiden Arthritis (RA) erwiesen.
Es ist insoweit eine Entwicklung zu einer personalisierten, patientenorientierten Präzisionsmedizin erkennbar: Die Wirkung von Adalimumab mit MTX war umso besser, je mehr HLA DBR1-Gen-Varianten der Patient hatte. Der Nachweis von mehreren „shared epitope“-Kopien spricht deshalb für eine frühzeitige Behandlung mit Adalimumab. Auf diese Weise können natürlich Gentests auch die Behandlungskosten senken und den Einsatz bestimmter Produkte in einer frühen Phase vertretbar machen. Hingegen gibt es keine Erkenntnis, dass ein Originator mit sämtlichen Biosimilars austauschbar wäre. Gerade bei Hochrisikopatienten ist deshalb die Austauschbarkeit eingeschränkt.
Diese Erkenntnisse vermehren sich, je mehr Biologika eingesetzt werden. Teilweise behalten dabei Register wie RABBIT die Biologika-Risiken im Blick.
Im Hinblick auf die Wirtschaftlichkeit bedeutet dies, dass ein Vertragsarzt möglicherweise die vorgegebenen Biosimilar-Quoten aus medizinischen Gründen nicht einhalten kann. Die Verordnung von Biosimilars wird von unterschiedlichen Faktoren beeinflusst (s. Abb. S. 19).
WICHTIG: Im Hinblick darauf schließt sich das Sozialrecht bei der Beurteilung der Wirtschaftlichkeit zivilrechtlichen Grundsätzen an. Nach den Vorgaben in § 9 der Arzneimittel-Richtline gilt ein Minimalprinzip, wenn das Therapieziel mit mehreren gleichwertigen Behandlungen und Strategien erreichbar wäre. In dieser Situation fordert die Rechtsprechung den Einsatz eines preisgünstigen Präparates (BSG v. 13.05.2015 – B 6 KA 8/14 R). Hingegen sind finanzielle Aspekte bei der Beurteilung der medizinischen Notwendigkeit der Heilbehandlung unbeachtet, wenn eine Gleichwertigkeit nicht gegeben ist (z. B. BGH v. 12.03.2003 – IV ZR 278/01).
Insoweit besteht dann eine einzelfallbezogene Prüfung, ob möglicherweise Anlass auch zur Verordnung des teuren Medikaments besteht (BSG v. 20.10.2004 – B 6 KA 41/03). Grundsätzlich ist ein Vertragsarzt zur Anwendung des effektivsten Mittels verpflichtet, zugleich soll er zivilrechtlich die schonendste Methode wählen. Es ist behandlungsfehlerhaft, wenn bei der medikamentösen Therapie erkennbare und vermeidbare Nebenwirkungen auftreten, in die der Patient nicht eingewilligt hat (z. B. BGH v. 15.03.2005 – VI ZR 289/03; sowie vom 17.04.2007 – VI ZR 108/06 zur Aufklärung bei Medikamentenwechsel). Deshalb rechtfertigen deutliche Nutzenvorteile auch höhere Kosten, wie dies z. B. bei Unverträglichkeiten, längerer Wirkung/seltenerer Einnahme, erheblicher Reduktion unerwünschter Arzneimittelwirkungen oder erheblichen Risikofaktoren in Behandlungsfällen vorstellbar ist (Grundsatzentscheidung BSG v. 31.05.2006 – B 6 KA 13/05 R).
Regionale Vereinbarungen versus Standards
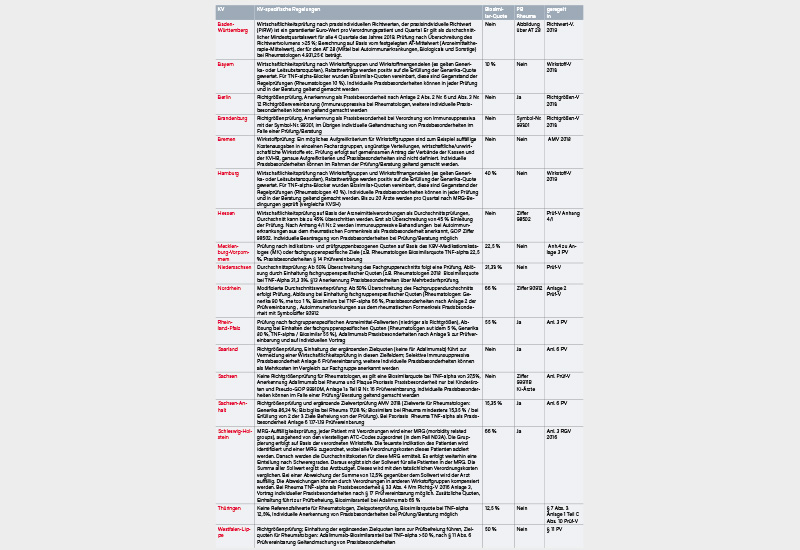
Die regionalen Kassenärztlichen Vereinigungen (KVen) halten für die Verordnung unterschiedliche Quoten vor, die für den jeweiligen Vertragsarzt unterschiedliche Bedeutungen haben (s. vorh. Tab.).
Bei den regionalen Vorgaben darf jedoch nach der Entscheidung des Bundessozialgerichts (BSG) vom 28.09.2016 – B 6 KA 43/15 R die Therapiefreiheit des Arztes nicht beschränkt werden. Der Arzt muss stets die Möglichkeit haben, z. B. auf Schwierigkeiten von Patienten im Umgang mit bestimmten Präparaten mit dem Wechsel auf ein anderes Medikament zu reagieren. Es ist unzulässig, die Zielwerte so zu bemessen, dass sie nur bei der Verordnung der preiswertesten Arzneimittel erreicht werden können.
Aus diesem Grund lassen die Prüfvereinbarungen auch stets die Möglichkeit zu, anderweitig zu verordnen. Ein Zwang zur Umstellung scheidet somit aus, bundesweite Standards sind also trotz der jeweiligen regionalen Vorgabe überall leistbar.
Prüfangst muss sich trotzdem nicht ausbreiten, zumal die Wirtschaftlichkeitsprüfungen insgesamt kaum noch messbar durchgeführt werden. Hintergrund sind formelle Probleme der Prüfgremien im Umgang mit den Rabattverträgen (die Rabatte werden gegenüber der Prüfungsstelle nicht offengelegt), so der Prüfung von AMNOG-bewerteten Produkten, die bereits aufgrund eines Rabatts als wirtschaftlich gelten. Da es keine Rechtssicherheit bei der Durchführung dieser Prüfungen gibt, hat dies nahezu zu einem Prüfstillstand geführt. Hinzu kommen erleichternde Regelungen wie der Grundsatz „Beratung vor Regress“, die dazu beitragen, dass eine Regressangst nicht die Triebfeder der Verordnung ist.
Rechtsanwalt Jörg Hohmann
Fachanwalt für Medizinrecht
Kanzlei für Medizinrecht
Prof. Schlegel Hohmann & Partner
Paul-Nevermann-Platz 5
22765 Hamburg